



























For medical device manufacturers, meeting compliance standards is an essential piece of the quality process. One of the most important of these necessary regulations is FDA 21 CFR Part 820, which ensures that medical devices are safe and effective. However, with fifteen different subparts, each with its own strict guidelines, remaining compliant with this standard can be difficult. In this article, we’ll outline each of 21 CFR Part 820’s subparts and give you a few tips on how to make compliance simple, including taking a proactive quality approach, performing internal audits, focusing on corrective action, and creating a strong document control system. Read on to learn more.
FDA 21 CFR Part 820 Requirements
Each of FDA 21 CFR Part 820’s fifteen subparts focuses on a particular area within medical device manufacturing and is labeled with a letter from A to O. Let’s go through what each one outlines, according to the FDA.
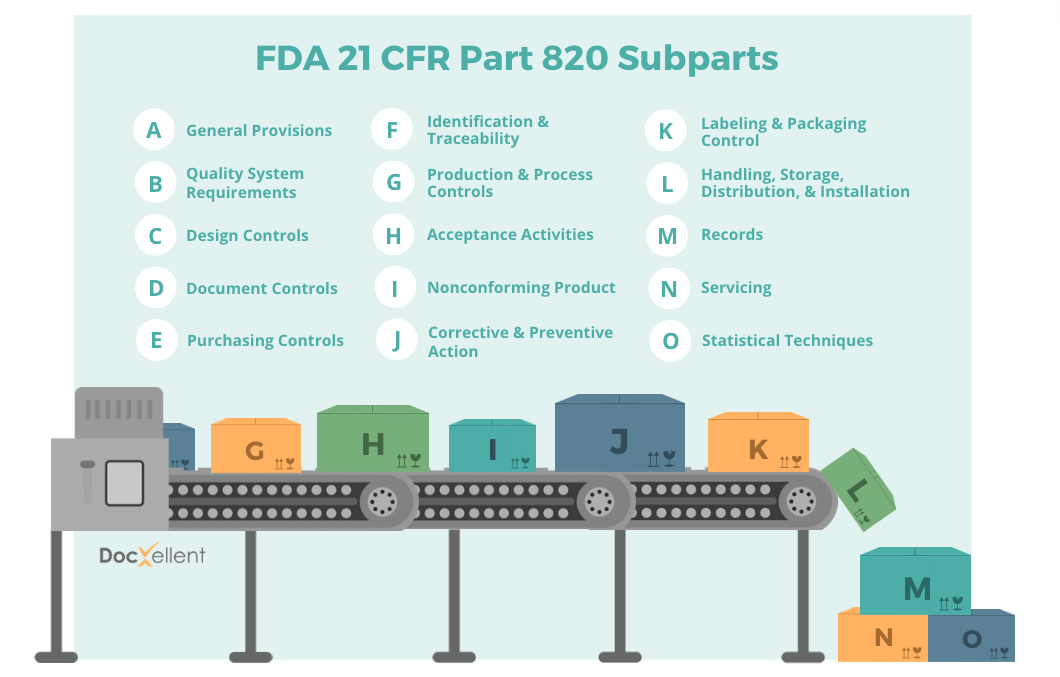
A: General Provisions
This subpart outlines which companies are required to adhere to the Federal Food, Drug, and Cosmetic Act. Essentially, 21 CFR part 820 applies to any medical device either manufactured in the US or imported in and intended for use in the US.
B: Quality system requirements
This section outlines the FDA’s requirements for a quality management system and what you should consider when implementing one. A quality management system should:
- Follow established and documented procedures
- Contain a quality plan for each part of the manufacturing process
- Enable management to review quality activities and report on its performance
C: Design controls
This subpart explains the controls your company should have in place for each section of your design process. Your quality management system should cover the following steps:
- Design and development planning
- Design input
- Design output
- Review
- Verification
- Validation
- Design transfer
- Changes
- History
D: Document Controls
The FDA outlines the methods your company is expected to control its documentation. Your processes need to include:
- Document approval and distribution
- Document change tracking
E: Purchasing Controls
Your company needs to thoroughly review potential suppliers or contractors to ensure they meet your outlined quality standards, keep records of your business relationship, and manage purchasing data.
F: Identification and Traceability
Any manufactured device must be marked with who made it, the make, and the model. This ensures that in the event of a recall or malfunction, the patient or healthcare provider can make a report and the issue can be easily resolved.
G: Production and Process Controls
This subpart states that each manufacturer must develop, conduct, control, and monitor production processes to ensure their products conforms to their specifications. This includes:
- Documenting instructions and standard operating procedures (SOPs)
- Implementing process controls for each step of the production process
- Ensuring all standards and regulations are complied with
- Validate and ensure the safety of all equipment and processes
- Adequately monitor and inspect your processes throughout
The full standard breaks down each aspect of these process controls: the surrounding environment, hygiene and handling controls, the use of equipment, and materials safety.
H: Acceptance Activities
Each manufacturer must establish and maintain procedures for acceptance activities: inspections, tests, and other verification activities.
I: Non-Conforming Product
In the case of a non-conforming medical device, you’ll need to address the identification, documentation, evaluation, segregation, and disposition of the product. Then, you’ll need to establish and maintain procedures for rework, including retesting and reevaluation to mitigate the issue.
J: Corrective and Preventive Action (CAPA)
Your company must establish and maintain procedures for implementing corrective and preventive action (CAPA). The procedures should include requirements for:
- Analyzing your sources of quality data to identify existing and potential causes of nonconforming products, or other quality problems
- Investigating the cause of nonconformities relating to products, processes, and the quality system
- Identifying the actions needed to correct and prevent the recurrence of nonconforming products and other quality problems
- Validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device
- Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems
- Ensuring that information related to quality problems or nonconforming products is disseminated to those directly responsible for assuring the quality of such products or the prevention of such problems
- Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review
K: Labelling and Packaging Control
Each manufacturer must establish and maintain procedures to control labeling activities. This includes:
- Label integrity
- Labeling inspection
- Labeling storage
- Labeling operations
- Control number
L: Handling, Storage, Distribution, and Installation
This subpart outlines the ways your company should work to ensure that mistakes don’t happen while the finished products are being handled and moved. Essentially, manufacturers should establish and maintain procedures to prevent mix-ups, damage, deterioration, contamination, or other adverse effects.
M: Records
Specific records need to be kept and made available for FDA inspection. These include:
- Device master record: includes information such as the device and production specifications; quality assurance procedures; labeling and packaging details, and installation, maintenance, and servicing instructions
- Device history record: key dates, quantities manufactured and distributed, and identification codes
- Quality system record: details the location of where procedures and activities are stored
- Complaint files: all details should be recorded, including the investigation and outcome of a complaint
N: Servicing
If servicing is a requirement for the medical devices your company manufactures, there should be detailed instructions for both validating and carrying this out. Records also need to be kept detailing when servicing takes place and who carried it out, including the specifics of what was done.
O: Statistical Techniques
Where appropriate, manufacturers shall establish and maintain procedures for identifying valid statistical techniques required for establishing, controlling, and verifying the acceptability of process capability and product characteristics.
How Can Your Company Simplify FDA 21 CFR Part 820 Compliance?
With fifteen complex subparts, compliance may seem like a daunting task. Here are four ways you can simplify this process.
- Implement a strong document control system
Under subpart D, your company is required to maintain quality records and appropriate controlled documentation throughout the manufacturing process. Therefore, to ensure compliance, it’s important to update your document control method and processes. With a strong document management software, you’ll have the ability to automate workflows, securely store your critical business documents, and have access to tools for collaboration, review and approvals, version control, specification management, corrective and preventative action, non-conformances, training, incidents, and reporting. There will be no risk of misplaced documentation or incorrect information, your data will be secure and accessible at any time.
- Perform internal audits
Another important part of part 820 compliance is internal auditing. Not only are these inspections a great way to prepare for an actual government audit, but the FDA expects medical device manufacturers to perform them to meet subpart B, which states: “Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established quality system requirements and to determine the effectiveness of the quality system.”
The key to this process is to set an audit schedule, stick to it, and set out to get the most value from it as you can. Audits are a way of assessing your resources, policies, and procedures. They provide a means for continuous improvement within your company. So, don’t treat internal audits as checkbox activities – perform them with purpose and use the information you gather to your advantage.
- Focus on corrective action
A huge part of Part 820 compliance is mitigating non-conformance. Organizations are required to create a detailed non-conformance plan and apply it to their monitoring, measurement, analysis, and improvement processes. To simplify this process, it’s important to streamline your corrective and preventive action (CAPA) method. You can do this in four steps:
- Reduce human error: Carefully define your CAPA process and set up logical workflows that eliminate the need for human involvement to keep your CAPAs consistent and beneficial.
- Be more proactive: Define and enforce CAPA processes in your organization carefully and consistently. Through building and monitoring processes into your business practices that help prevent issues from happening in the first place, your CAPA process will continue growing in efficiency and practicality.
- Set realistic deadlines: Set up a task management workflow that assigns a manager as the owner of each CAPA task, notifies them of all the data collected so far, and keeps them up to date on the status. This way, when unforeseen problems arise, managers close to the problem know what it takes to deliver on time and can adjust accordingly.
- Evaluate and modify your process: Without measuring your quality results and the way your CAPA process is performing, there's not much use in attempting preventive actions. So, it is important to make sure that you are always collecting data about how problems occur and how you solve them.
For a more in-depth discussion on streamlining your CAPA process, check out this guide: How to Optimize the CAPA Workflow to Increase Quality.
- Take a proactive approach to quality management
As stated in subpart B, your company is expected to establish a suitable Quality Management System (QMS), provide the resources necessary for a QMS, and establish and maintain training records. Part of this process is taking a proactive approach to your quality management process and creating a culture of quality throughout your company.
To do so, find a QMS that allows you to do the following:
- Automate the tracking of your employees’ training records
- Meet training requirements through established training rosters
- Internal & external training events and configurable training reports
- Manage, schedule, and record all audit activities
- Audit reporting that compiles compliance suggestions and recommendations into a consolidated report
With these features in place, your company will cultivate a proactive quality process and meet the QMS requirements under FDA 21 CFR Part 820.
It’s a lot of work to make sure you’re keeping up with FDA standards. With ever-changing regulations and strict audit processes, it’s hard to know if your procedures meet their expectations. However, if you follow the four tips we’ve outlined today, you can simplify this process and take the stress out of compliance.
When you want to streamline your quality control management and easily meet FDA 21 CFR Part 820 standards, you can depend on ENSUR QMS software. To learn more about how ENSUR can benefit your quality control efforts, contact us now or Request A Demo.